

What Is Hepatolenticular Degeneration?
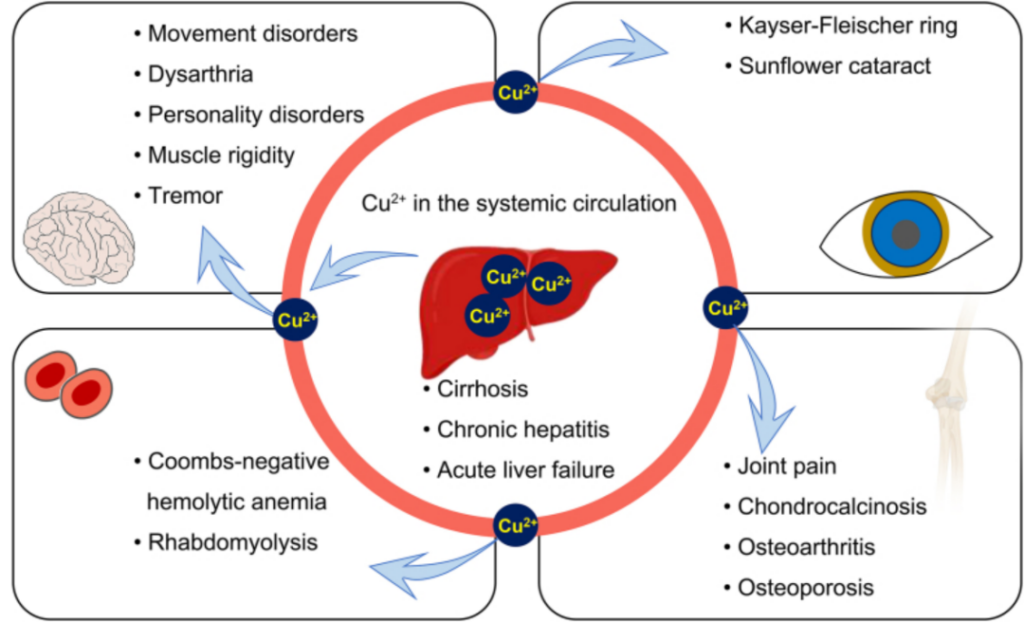
Hepatolenticular Degeneration, also known as Wilson Disease (WD), is a rare autosomal recessive genetic disorder caused by mutations in the ATP7B gene, which leads to abnormal copper accumulation in organs including the liver, brain and cornea. Most WD patients develop symptoms at a young age, with hallmark clinical manifestations comprising extrapyramidal signs, liver cirrhosis, corneal Kayser-Fleischer (K-F) rings, low serum ceruloplasmin levels (<50 mg/L), and elevated 24-hour urinary copper excretion (>100 mg/L). The global prevalence of WD ranges from 1/30,000 to 1/50,000; its incidence is higher in China than in Western populations, with an onset age spanning from 1 to 72 years old.
Pathogenesis
WD arises from mutations in the ATP7B gene, which encodes the ATP7B copper-transporting ATPase located on the long arm of chromosome 13. Expressed in hepatocytes, ATP7B exerts dual regulatory functions on copper homeostasis: under low intracellular copper concentrations, ATP7B localizes to the Golgi apparatus to facilitate holoceruloplasmin synthesis. When hepatic copper levels rise, ATP7B relocates to shuttle excess copper for biliary excretion. Additionally, ATP7B sequesters surplus cellular copper within vesicles and transports these vesicles to the cell membrane for extracellular copper efflux.
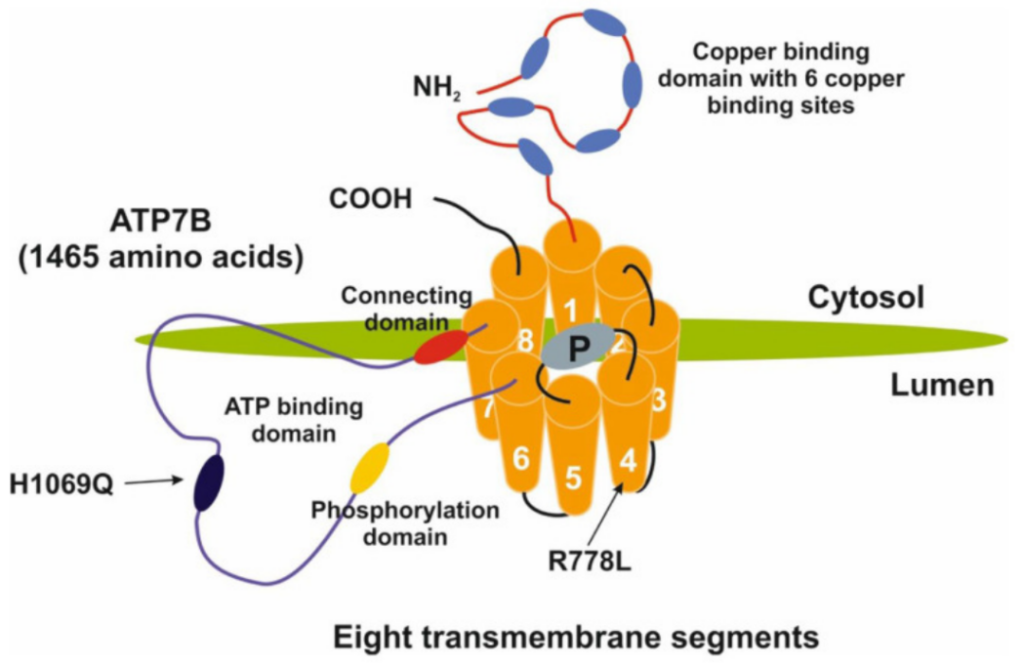
Pathogenic ATP7B mutations impair ATP7B protein function, disrupting ceruloplasmin biosynthesis and reducing biliary copper excretion, thereby triggering massive copper retention in the liver. Once the liver’s copper storage capacity is saturated, copper leaks into the bloodstream and deposits in extrahepatic organs such as the brain, cornea and kidneys, inducing oxidative damage and irreversible organ dysfunction.
ATP7B mutation spectra exhibit prominent ethnic and geographic disparities. Over 800 distinct pathogenic variants have been documented, including missense, nonsense, insertion/deletion and splice-site mutations.
- Caucasian populations: The p.H1069Q (c.3207C>A) variant predominates, accounting for 37%–63% of mutant alleles in European cohorts.
- Asian populations: Common variants include p.R778L (c.2333G>T), p.P992L (c.2975C>T) and p.T935M, with respective allele frequencies of 28.7%, 9.3% and undetermined prevalence.
Gene Editing & Therapeutic Strategies
- Gene Editing Technology Gene editing enables targeted correction of pathogenic ATP7B mutations to restore physiological copper transport function, with promising preliminary outcomes validated in preclinical animal models.
- AAV Vector-Mediated Gene Therapy Adeno-associated virus (AAV) vectors deliver wild-type ATP7B transgenes to hepatocytes; preclinical studies have validated restored copper metabolic homeostasis. In December 2024, the MWAV201 clinical trial led by Professor Jiangao Fan, Director of the Gastroenterology Department at Xinhua Hospital Affiliated to Shanghai Jiao Tong University, completed the first drug administration in Wilson Disease patients, with well-tolerated treatment and no adverse events observed to date. On February 6, 2025, GC310, an AAV injectable gene therapy independently developed by Beijing Genecradle Biotechnology Co., Ltd., received implied approval for clinical trials from the National Medical Products Administration (NMPA).
Research Mouse Models for Wilson Disease
- Atp7b<sup>-/-</sup> Knockout Mice Gene-targeted ablation of murine Atp7b abolishes functional ATP7B protein, inducing severe hepatic copper overload and recapitulating copper metabolic dysregulation characteristic of human WD.
- TX-J Mice Carrying the c.2135G>A point mutation in Atp7b that causes the p.G712D amino acid substitution, this strain recapitulates core WD phenotypes including hepatic copper accumulation, mitochondrial dysfunction and inflammatory responses.
- p.R778L Knock-In Mice The c.2333G>T variant (p.R778L) recapitulates the most prevalent Asian WD mutation; these mice display neurological deficits including impaired motor coordination and cognitive dysfunction, accompanied by elevated pro-inflammatory cytokine expression in the central nervous system.
- p.H1069Q Knock-In Mice Engineered to harbor the c.3207C>A (p.H1069Q) variant prevalent in European populations, this model serves as a critical tool for dissecting WD pathogenic mechanisms and developing targeted therapeutics for Western patient cohorts.
MingCeler Biotech Supports Gene Therapy Development
Gene therapy brings new therapeutic hope for rare diseases, yet its development and efficacy validation rely heavily on standardized animal models. Leveraging our proprietary TurboMice™ platform, MingCeler Biotech has generated a comprehensive portfolio of rare disease mouse models. TurboMice™ overcomes two major bottlenecks of traditional model generation: prolonged breeding cycles and low success rates for complex genetically modified strains. The platform supports precise editing of nearly any target genomic locus and generates fully homozygous gene-edited mice directly from embryonic stem cells in as little as 2 months.
MingCeler Biotech provides custom Wilson Disease mouse models tailored to client research demands, including Atp7b<sup>-/-</sup> knockout mice, TX-J mice, p.R778L knock-in mice and p.H1069Q knock-in mice. Contact our technical team for inquiries!
References
[1] Immergluck J, Grant LM, Anilkumar AC. Wilson Disease. [Updated 2025 Apr 3]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441990/
[2] Xiong X, Gao C, Meng X, Liu A, Gong X, Sun Y. Research progress in stem cell therapy for Wilson disease. Regen Ther. 2024 Mar 15;27:73-82. doi: 10.1016/j.reth.2024.03.005. PMID: 38525238; PMCID: PMC10959646.
[3] Kucinskas L, Jeroch J, Vitkauskiene A, Sakalauskas R, Petrenkiene V, Kucinskas V, Naginiene R, Schmidt H, Kupcinskas L. High frequency of the c.3207C>A (p.H1069Q) mutation in ATP7B gene of Lithuanian patients with hepatic presentation of Wilson’s disease. World J Gastroenterol. 2008 Oct 14;14(38):5876-9. doi: 10.3748/wjg.14.5876. PMID: 18855987; PMCID: PMC2751898.
[4] Malipati A, Kaidiriya K, Xu L, Sun X. Research advances in the pathogenesis, phenotype-genotype relationship, and pharmacotherapy of hepatolenticular degeneration. Department of Infectious Diseases, The First Affiliated Hospital of Xinjiang Medical University, Urumqi 830000, China.
[5] https://www.bj-genecradle.com/news/276
[6] Penning LC, Berenguer M, Czlonkowska A, Double KL, Dusek P, Espinós C, Lutsenko S, Medici V, Papenthin W, Stremmel W, Willemse J, Weiskirchen R. A Century of Progress on Wilson Disease and the Enduring Challenges of Genetics, Diagnosis, and Treatment. Biomedicines. 2023 Feb 1;11(2):420. doi: 10.3390/biomedicines11020420. PMID: 36830958; PMCID: PMC9953205.
[7] Yang YL, Wei TH, Yang WM, et al. Research advances in animal models of hepatolenticular degeneration. J Clin Hepatol. 2022, 38(5): 1169-1174. DOI: 10.3969/j.issn.1001-5256.2022.05.041.
[8] Dong J, Xiang G, Xia X, et al. Aberrant copper metabolism and hepatic inflammation cause neurological manifestations in a mouse model of Wilson’s disease. J Neuroinflammation 21, 235 (2024). https://doi.org/10.1186/s12974-024-03178-5